Ketonuria estas la ĉeesto en la urino de cetonaj korpoj, nome acetono
La mekanismo de ketonuria.
En sana homo, karbonhidratoj, lipidoj kaj iuj proteinoj estas oksiditaj al karbona dioksido kaj akvo. En iuj patologiaj kondiĉoj, precipe kun diabeto, produktado de insulino malpliiĝas. En la hepato reduktiĝas rezervoj de glicogeno, multaj korpaj histoj estas en energia malsato. Sub ĉi tiuj kondiĉoj, la procezoj de oksidigo de proteinoj kaj lipidoj en la hepato estas aktivigitaj, sed manko de glukogeno kondukas al ilia nekompleta oksido kaj amasiĝo en la sango de neoksidigitaj produktoj de lipida kaj proteina metabolo - ketonaj korpoj. Pro la amasiĝo de cetonaj korpoj en la sango (ketonemio), la pH de la sango translokiĝas al la acida flanko. Ĉi tiu kondiĉo nomiĝas akidozo. La urino de tia paciento havas akran acidan reagon kaj odoras al acetono.
Starvation, la uzo de ĉefe proteinoj kaj grasaj manĝaĵoj, la ekskludo de karbonhidratoj el la dieto kondukas al pliigita formado de ketonaj korpoj kaj ilia ekskrecio en la urino.
En frua aĝo, ketonuria estas pli ofta ol ĉe plenkreskuloj kaj ne havas klinikan signifon. Ĉi tiu fenomeno interesas la infankuraciston kun "aketonemia vomado" asociita kun eraroj en la dieto.
Ketonuria en la postoperacia periodo estas pro proteina rompo pro kirurgia traŭmato.
Ĉe plenkreskuloj, ketonuria okazas en severaj formoj de diabeto kaj havas grandan diagnozan valoron. En infanoj, ĝi povas esti kun diversaj malsanoj, pro la kapablo de karbonhidrata metabolo. Sekve, eĉ malgrandaj eraroj en la dieto, precipe en ĉeesto de akra infekto, nerva ekscitiĝo, troa laboro ktp. povas konduki al ketosis. Ketonuria en frua infanaĝo povas esti observata kun toksikozo, longaj gastrointestinalaj malordoj, disenterio kaj aliaj malsanoj. Ĉe novnaskitoj, kresko de urinaj cetonoj estas preskaŭ ĉiam kaŭzita de subnutrado. Ketonuria, observata en infektaj malsanoj - skarlata febro, gripo, tuberkula meningito kaj enaksiĝo, estas malĉefa signo, traira kaj ne havas grandan diagnozan valoron.
Ketonuria en diabeto disvolviĝas pro pliigita ketogenezo kaj neplena ketolizo. Pliigita ketogenezo kondukas al pliigita mobilizado de grasoj el adiposa histo, malpliiĝo en la formado de oksalacetato en la ciklo de Krebs, kaj malpliigo de la biosintezo de grasaj acidoj. En severa diabeto, akompanata de damaĝo al la rena histo (loko por rompo de ketonoj), plia malobservo de ketolizo okazas.
La foresto de glucosuria en ĉeesto de ketonuria ekskludas diabeton.
Kutime, cetonaj korpoj formiĝas en malgranda kvanto el la fina produkto de karbonhidrata kaj lipida metabolo - acetil-CoA (C2-korpoj) per acetoacetil-CoA kaj estas preskaŭ tute neŭtraligitaj.
En diabeto mellitus, la mobilizado de lipidoj pliiĝas kun la formado de granda kvanto da acetila CoA (C)2korpo). samtempe, pro malobservo de karbonhidrata metabolo, malpliigo de la formado de oksalacetato, kun kio C2-korpoj estas inkluzivitaj en la ciklo de Krebs kaj brulas ĝis karbona dioksido kaj akvo. Krome, rezulte de pliigita lipida rompo, la inversa biosintezo de C estas blokita.2korpo en grasajn acidojn. Tiel, granda kvanto da C akumuliĝas2-unuoj, kio kaŭzas produktadon de granda kvanto da CoA, kaj sekve acetona, acetoacetika kaj beta-hidroksibutria acido, kiuj eltiras en la urino.
Ketonaj korpoj
Ĉi tiuj estas mezaj kadukaj produktoj, kiuj formiĝas dum hepofunkcio. Kutime, metabolo eksponas cetonajn korpojn al plia degenero.
La rezervoj de glukozo de la korpo troviĝas en korpa graso, tial ketonuria estas forta manko de karbonhidratoj. Ketonaj korpoj estas bonega provizanto de energio. En ĉi tio eĉ grasaj acidoj restas malantaŭ ili. Tial, kiam la cerbo aŭ koro malhavas fosforajn acidojn, la korpo tuj produktas cetonajn korpojn rapide.
Kiuj estas la kialoj
Ketonuria estas la urina enhavo de cetonaj korpoj super normala.
En sana homa korpo, metabolo bone ekvilibrigas. Kio povas kaŭzi ketonurion?

- La superregado de proteino kaj graso en la dieto. Pro malpliigo de karbonhidratoj en manĝaĵo, ĉeloj mankas nutraĵon. En ĉi tiu fono disvolviĝas ketonuria. Ĉi tio estas reago de la korpo al malekvilibro en la dieto.
- Dietoj kaj misuzo de malsato povas deĉenigi la aperon de ketonaj korpoj en la urino. Eraroj en la elekto de dietoj kondukas al rapida rompo de grasoj, dum la nombro de enzimoj pliigas. Ketonuria estas ankaŭ la apero de acetono en la sango. Kutime, se homo malsatas dum pli ol ses tagoj, la enhavo de cetonaj korpoj en la homa korpo pliiĝas super normalo.
- Anestezio por kirurgio.
- Diabeto mellitus. Ĉi-kaze ketonuria aperas de tempo al tempo. Ketonaj korpoj sole ne kaŭzas la malsanon. En diabeto mellitus, aldona kurso de karbonhidratoj kaj insulino estas preskribita.
- Deshidratigo. Ĝi okazas kiam la korpo varmigas aŭ estas komplikaĵo de diabeto.
- Hepato-malsano aŭ severaj infektaj malsanoj (t.e. disenterio).
- La disvolviĝo de tumoroj en la digesta vojo.
- Malordoj en la pankreato.
- Veneniĝo per alkoholo aŭ per tiaj kemiaj komponaĵoj kiel fosforo, plumbo.
- Damaĝo al la centra nerva sistemo, nerva ekscito.
Memoru, ke la odoro de acetono kiam spirado aŭ urinado estas signalo por vidi kuraciston. Ĝi ankaŭ estas kialo por ŝanĝi vivstilon kaj nutran ekvilibron.
Atentu infanojn
La kliniko ofte konsultiĝas se la infano havas vomadon, la odoron de acetono. Aŭ tia odoro aperis en la urino. Kaj kvankam ĝi estas signo de ketonuria, ĝi ne nepre estas simptomo de grava malsano.

Ofte, ĉio estas klarigita per neperfekta metabola sistemo. La kaŭzo de ketonuria en infanoj estas kondiĉo en kiu elspezas signifa kvanto da energio. Ĝi kutime okazas kiam:
- emocia malordo
- pliigita fizika streĉo,
- malekvilibra dieto
- malvarmo
Fakte, la korpo de la infano ne havas grandajn butikojn de glukogeno, tial aktiva rompo de grasoj okazas kaj signoj de ketonuria estas observataj.
Gravedeco
Dum naŭ monatoj de gravedeco, virinoj ofte devas fari testojn de urino. Ĉi tio estas necesa por ne maltrafi la plej etan devion de la normo en la korpo por la tuta periodo de gestado. Ja ĉiuj organoj havas grandan ŝarĝon. Kion signifas ke cetonaj korpoj troviĝas en urino?
Kutime ilia malgranda enhavo estas la normo. La plej simpla analizo pasos la rapidan teston. Vi devas mallevi la testan strion en la urino.
Negativa testo estas konsiderata normala dum gravedeco aŭ la nombro de cetonoj estas minimuma. Se la prova valoro estas de 15 ĝis 160 mg / dl - tio kaŭzas zorgon.
Dum gravedeco, ketonuria estas alarmiga simptomo. Ĝi aperas kun frua toksikozo. La veneniĝo de la korpo kaj tial la feto kun acetono, komplikas la kurson de gravedeco.

Se gravedulino havas gravan hormonan malsukceson, ŝi riskas.
Pliiĝo de la ketonoj en la urino de novnaskito estas pro nesufiĉa nutrado aŭ nutraj eraroj.
Simptomoj de Ketonuria
Se la nivelo de ketonoj en la korpo pliigis signife, tiam ketonuria manifestos sin:
- elĉerpanta somnolo
- odoron de acetono el la buŝo,
- urina acetona odoro
- analizo montros altan nivelon de blankaj globuloj en la sango,
- biokemia sangotesto montros malaltan glukozon,
Se ketonuria provokis salton en la sangon de acetono, tiam povas okazi kriza acetono.
Signifa kresko de acideco en ĉeloj povas serioze damaĝi internajn organojn. En ĉi tiu kazo, protekta reago estas lanĉita - vomado.
Simptomoj de ketonuria en infanoj:
- plendoj de abdomina doloro
- plendoj pri kapdoloroj
- la infano estas laca, letargia,
- plendoj de naŭzo
- vomado
- levante la temperaturon al 39 ° C,
- rifuzo de manĝaĵo
- odoras kiel acetono
- pligrandigita hepato
Ĉu ketonuria povas esti detektita?
Nur kemia analizo povas detekti pliigon de la nivelo de cetonaj korpoj en la korpo. La laboratorio tuj starigos la normon de la cetonaj korpoj en ĝi.
En moderna medicino, ketonuria estas detektita:
- Langa testo,
- Leĝa testo
- Specimena Lestrado,
- modifita specimeno de Rothera,
- rapidaj provoj

Rapidaj provoj kompreneble estas la plej oftaj. Ilia ago baziĝas sur kemia reago, kies rezulto estas videbla preskaŭ tuj. Oni nur metas la testan strion en la urino aŭ faligas ĝin sur testan tablojton. En kazo de pozitiva reago, la testoj fariĝas purpuraj. La brilo de la koloro permesas juĝi per speciala kolora skalo pri kiom multe superas la normo de cetonaj korpoj.
Internacia Klasifiko de Malsanoj
La Internacia Statistika Klasifiko de Malsanoj kaj Rilataj Sanproblemoj (ICD) estas referenca gvidilo per kiu materialoj kompareblas internacie. Kun ĝia helpo, diversaj metodikaj aliroj estas kombinitaj. Statistikoj pri malsanoj estas akumulataj kaj klasigitaj. Ĉiuj dek jaroj, IBC estas reviziita de komisiono de la Monda Organizaĵo pri Sano. Por klasifiki kaj analizi la tutan amason de amasigitaj rezultoj el la vidpunkto de statistikoj, ĝi estas tradukita al alfanombraj kodoj. En tia laboro oni uzas la disvolvitan ICD. Hodiaŭ ĝi respondas al la ICD-10. Ĝi enhavas 22 klasojn (sekcioj).
Laŭ la adresaro ICD, ketonuria havas la kodon R82.4.
Ketonuria Antaŭzorgo kaj Dieto
Por preventado de ketonuria necesas:
- manĝi ĝuste
- konduki sanan vivstilon:
- kiel eble plej multe esti en la freŝa aero
- modera ekzercado
- Ne komencu kronikajn malsanojn.

En kronika malsano kiel diabeto, kuracisto konsultas sisteme. De tempo al tempo necesas fari espriman teston.
Pozitiva reago indikas patologion en la korpo. Atentu la signalon! Urĝa apelacio al la kliniko rapide redonos la korpon al normalo. Sen foriri de la dieto preskribita de la kuracisto, vi povas plirapidigi resaniĝon.
Ketonuria dieta principoj:
- ne manĝu manĝaĵojn altajn proteinojn kaj grasojn,
- regula konsumado de karbonhidratoj
- trinku pli da akvo kaj sodaj solvoj (kun diabeto - insulino).
La menuo devus inkluzivi: boligitajn bovaĵon kaj kuniklon, diversajn vegetalajn supojn, malaltan grasan fiŝon. Utilaj cerealoj sen oleo, legomoj kaj fruktoj. Oni rekomendas trinki pli da sukoj, fruktaj trinkaĵoj, kompotoj.
Ekskludi de menuo:
- grasa viando
- spicaj kondimentoj
- dolĉa
- citrusaj fruktoj
- bananoj
- fungoj
- rapidmanĝaĵo.
Fokuso sur kuracado
Ketonuria ne devas esti traktata kiel aparta malsano. Ŝi estas nur konsekvenco. Oni devas ekskludi la kialojn, kiuj kaŭzis ĝin. Unue necesas kompleta ekzameno. La ĝusteco de la diagnozo kaj la starigo de la kaŭzo de ketonuria garantias sukcesan kuracadon.
Kuracistoj donas iujn konsilojn:
- Se vi estas troa, vi devas aranĝi de tempo al tempo fastojn.
- Se la analizo rivelis kreskon de urinaj cetonoj, aĉetu testojn por uzi ilin hejme.

- La urino devas esti kolektita por analizo ne pli ol kvar horojn antaŭ liverado.
- La infano povas ebriiĝi per alkala trinkaĵo ĉiujn 10-15 minutojn en malgrandaj porcioj. Aktivigita karbo, enterosgel helpos malplenigi la digestan tracton.
- Kun venonta vomado, estas utile preni frakcian trinkaĵon. Voki ambulancon.
Kiel trakti ketonurion, estas konsiloj en popola medicino.
- Kun ofta vomado, trinku mineran akvon, sekan fruktan kompoton, glukozan solvon. Unu kulero en dek minutoj.
- Metu malamikon hejme. Unue, akvon je ĉambra temperaturo, poste varme, en kiu aldoniĝas kulero da sodeto.
- Prenu trinkaĵon: solvu 2 tbsp en 1 litro da akvo. mielo, verŝu la sukon de unu citrono. Trinku 1 tbsp. ĉiun 15 minutojn.
- Recepto por natria solvo: solvu 1 cucharadon da natrio en 250 ml da akvo. Trinku 1 tsp. ĉiun 10 minutojn.
- Prenu ordenojn de trankviligaj herboj.
- Por rapidigi la eliminon de toksinoj el la korpo, manĝu iom post iom. Pli bone estas manĝi nur biskvitojn.
Traktado dependas de la individuaj trajtoj de la korpo. Ĝi devas esti strikte kontrolata de la ĉeestanta kuracisto.
76. Kolesterolo. Manieroj de eniro, uzo kaj ekskrecio de la korpo. Seruma kolesterolo. Biosintezo de kolesterolo, ties etapoj. Reguligo de sintezo.
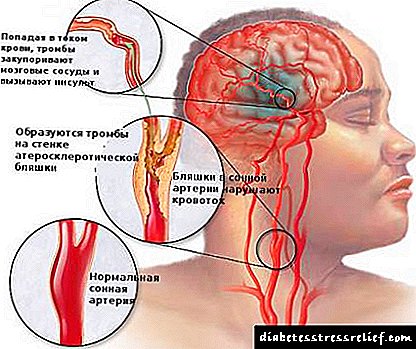
Kolesterolo estas steroido specifa al bestaj organismoj. Ĝi estas sintezita en multaj homaj histoj, sed la ĉefa loko de sintezo estas la hepato. En la hepato, pli ol 50% de kolesterolo estas sintezita, en la malgranda intesto - 15-20%, la resto de la kolesterolo estas sintezita en la haŭto, suprena cortico kaj gonadoj. Ĉirkaŭ 1 g da kolesterolo estas sintezita ĉiutage en la korpo, 300-500 mg estas ingestitaj per manĝaĵo (Fig. 8-65). La kolesterolo plenumas multajn funkciojn: ĝi estas parto de ĉiuj ĉelaj membranoj kaj influas iliajn propraĵojn, servas kiel komenca substrato en la sintezo de galaj acidoj kaj steroidaj hormonoj. Antaŭdirintoj en la metabolan vojon de sintezo de kolesterolo ankaŭ transformiĝas en ubiquinono, komponanto de la spira ĉeno kaj dolikolo, kiu okupiĝas pri la sintezo de glicoproteinoj. Pro sia hidroksil-grupo, kolesterolo povas formi esterojn kun grasaj acidoj. Etherifita kolesterolo predominas en la sango kaj estas konservita en malgrandaj kvantoj en iuj specoj de ĉeloj, kiuj uzas ĝin kiel substraton por la sintezo de aliaj substancoj. Kolesterolo kaj ĝiaj esteroj estas hidrofobaj molekuloj, do ili estas transportataj per sango nur kiel parto de diversaj specoj de drogoj. La interŝanĝo de kolesterolo estas ege kompleksa - nur por ĝia sintezo necesas ĉirkaŭ 100 sinsekvaj reagoj. Entute ĉirkaŭ 300 malsamaj proteinoj okupiĝas pri metabolo de kolesterolo. Malordoj de kolesterolo-metabolo kondukas al unu el la plej oftaj malsanoj - aterosklerozo. La morteco pro la efikoj de aterosklerozo (miokardia infarkto, frapo) kondukas en la ĝenerala strukturo de la morteco. Aterosklerozo estas "poligena malsano", t.e. multaj faktoroj okupiĝas pri ĝia evoluo, la plej gravaj el ili estas heredaj. La amasiĝo de kolesterolo en la korpo kondukas al disvolviĝo de alia ofta malsano - malsano biliara.
A. Sintezo de kolesterolo kaj ĝia regulado
Reagoj de sintezo de kolesterolo okazas en la citosol de ĉeloj. Ĉi tiu estas unu el la plej longaj metabolaj vojoj en la homa korpo.
Fenilketonuria

Fenilketonuria - hereda malobservo de metabola aminoacido pro manko de hepataj enzimoj implikitaj en la metabolo de fenilalanino al tirozino. La fruaj signoj de fenilketonuria estas vomado, letargio aŭ hiperaktiveco, odoro de muldilo el urino kaj haŭto, malfrua psikomotoro-evoluo, tipaj malfruaj signoj inkluzivas oligofrenion, fizikan disvolvan malfruon, konvulsiojn, ekzememajn haŭtajn ŝanĝojn, ktp. Kribrado de novnaskitoj pro fenilketonuria efektiviĝas eĉ en patrina hospitalo, postaj diagnozoj inkluzivas molekulan genetikan testadon, determinon de sanga fenilalanina koncentriĝo, biokemia analizo de urino, EEG, kaj MRI de la cerbo. La kuracado de fenilketonuria estas sekvi specialan dieton.

Ĝenerala informo
Fenilketonuria (malsano de Felling, fenilpruviĉa oligofrenio) estas kungena, genetike determinita patologio, karakterizita de difektita hidroksilado de fenilalanino, la amasiĝo de aminoacidoj kaj ĝiaj metabolitoj en fiziologiaj fluidoj kaj histoj, sekvata de severa damaĝo al la centra nerva sistemo. Phenylketonuria estis unuafoje priskribita de A. Felling en 1934; ĝi okazas kun ofteco de 1 kazo po 10.000 novnaskitoj. En la novnaska periodo, fenilketonuria ne havas klinikajn manifestaĵojn, tamen la konsumado de fenilalanino kun manĝaĵo kaŭzas la manifestiĝon de la malsano jam en la unua duono de la vivo, kaj poste kondukas al severaj evoluaj malordoj de la infano. Tial la antaŭ-simptoma detekto de fenilketonuria ĉe novnaskitoj estas la plej grava tasko de neonatologio, pediatrio kaj genetiko.
Kaŭzoj de Phenylketonuria
Fenilketonuria estas aŭtosomal recesiva heredaĵmalordo. Ĉi tio signifas, ke por la disvolviĝo de klinikaj signoj de fenilketonuria, la infano devas heredi unu difektan kopion de la geno de ambaŭ gepatroj, kiuj estas heterozigaj portantoj de la muta geno.
Plej ofte, la disvolviĝo de fenilketonuria estas kaŭzita de mutacio en la geno kodanta la enzimon fenilalanino-4-hidroksilase situantan sur la longa brako de la kromosoma 12 (lokuso 12q22-q24.1). Ĉi tiu estas la tiel nomata klasika fenilketonuria, kiu reprezentas 98% de ĉiuj kazoj de la malsano. Hiperfenilalaninemio povas atingi 30 mg% kaj pli. Se ĝi ne traktas, ĉi tiu fenilcetonuria varianto estas akompanata de profunda mensa prokrasto.
Krom la klasika formo, atipikaj fenilketonuria distingiĝas, procedante kun la samaj klinikaj simptomoj, sed ne kapablaj korekti per dieta terapio. Ĉi tiuj inkluzivas fenilketonuria tipo II (manko de dehidroterterina reduktasa), fenilketonuria tipo III (manko de tetrahidrobiopterino) kaj aliaj pli maloftaj variantoj.
La probablo naski infanon kun fenilketonuria pliiĝas kun proksimaj geedzecoj.
Patogenesis de fenilketonuria
La klasika formo de fenilketonuria baziĝas sur la nesufiĉo de la enzimo fenilalanino-4-hidroksilase implikita en la konvertiĝo de fenilalanino al tirozino en mitokondria hepatocito. Siavice, la derivaĵo de tirozino estas la komenca materialo por la sintezo de catecolaminoj (adrenalino kaj norepinefrino), kaj diiodotrosino por la formado de tiroksino. Krome la formado de melanina pigmento estas rezulto de fenilalanina metabolo.
Heredeca manko de la fenilalayin-4-hidroksilase-enzimo en fenilcetonuria kondukas al malobservo de la oksidado de fenilalanino el manĝaĵo, kio rezultigas ĝian koncentriĝon en la sango (fenilalaninemio) kaj cerebrospinal fluido pliigas signife, kaj la nivelo de tirozino malkreskas sekve. Ekscesa fenilalanino estas forigita per pliigita urina ekskrecio de siaj metabolitoj - fenilpruviĉa acido, fenilmilaktika kaj fenilacetika acido.
Interrompo de aminoacida metabolo estas akompanata de difektita mielinigo de nervaj fibroj, malpliiĝo de la formado de neurotransmisiloj (dopamino, serotonino, ktp.), Ekigante patogenetajn mekanismojn de mensa prokrasto kaj progresema demenco.
Simptomoj de fenilketonuria
Novnaskitoj kun fenilketonuria ne havas klinikajn signojn de la malsano. Tipe, la manifestiĝo de fenilcetonuria en infanoj okazas en la aĝo de 2-6 monatoj. Kun la komenco de nutrado, la proteino de patrina lakto aŭ ĝiaj anstataŭantoj komencas eniri la korpon de la bebo, kio kondukas al disvolviĝo de la unuaj, nespecifaj simptomoj - letargio, foje maltrankvilo kaj hiper-ekscitemo, regurgitado, muskola distonio, konvulsia sindromo. Unu el la fruaj patognomonaj signoj de fenilketonuria estas konstanta vomado, kiu ofte estas erare konsiderata kiel manifestiĝo de pora historia stenozo.
Je la dua duono de la jaro, la malfruo de infano en psikomotora disvolviĝo fariĝas rimarkinda. La infano fariĝas malpli aktiva, indiferenta, ĉesas rekoni amatojn, ne provas sidiĝi kaj stariĝi sur la piedoj. La eksternorma konsisto de urino kaj ŝvito kaŭzas karakterizan odoron de "muso" (la odoro de muldilo) emananta de la korpo. Ofte estas senŝeligado de la haŭto, dermatito, ekzemo, sklerodermo.
En infanoj kun fenilketonuria, kiuj ne ricevas kuracadon, mikrocefalia, prognatio, poste (post 1,5 jaroj) dentado, emajla hipoplasio estas detektitaj. Oni rimarkas malfruon en parolad-disvolviĝo, kaj antaŭ 3-4 jaroj profunda oligofrenio (idioteco) kaj preskaŭ kompleta foresto de parolado estas detektitaj.
Infanoj kun fenilketonuria havas displastikan fizikon, ofte kungenitalajn kor-difektojn, aŭtonomajn disfunktojn (ŝvito, acrocianosis, arteria hipotensio) kaj suferas estreñimiento. Fenotipaj trajtoj de infanoj suferantaj de fenilketonuria inkluzivas malpezan haŭton, okulojn kaj harojn. Infano kun fenilketonuria estas karakterizita per specifa pozicio de "tajloro" (supraj kaj malsupraj membroj fleksitaj ĉe la artikoj), mano-tremado, vobla, mina marŝado kaj hiperkinezo.
La klinikaj manifestaĵoj de tipo II-fenilketonuria estas karakterizitaj de severa grado de mensa malfruiĝo, pliigita iritiĝemo, kaptiloj, spastic tetraparesis kaj tendona hiperrefleksio. La progresado de la malsano povas konduki al morto de infano en aĝo de 2 ĝis 3 jaroj.
Kiam fenilketonuri tipo III disvolvas triadon de signoj: mikrocefalio, oligofrenio, spasta tetraparesis.
Diagnozo de fenilketonuria
Nuntempe, la diagnozo de fenilketonuria (same kiel galaktosemio, kongenita hipotiroidismo, adrenogenita sindromo kaj kistika fibrozo) estas parto de la neonatala kribra programo por ĉiuj ĵus naskitaj.
Kribrilo-testado estas farata dum 3-5 tagoj da vivo de plenlonga kaj 7 tagoj de vivo de antaŭtempa bebo prenante specimenon de kapilara sango sur speciala papera formo. Se hiperfenilalanemio estas detektita, pli ol 2,2 mg% de la infano estas raportita al infana genetiko por reekzameno.
Por konfirmi la diagnozon de fenilketonurio, la koncentriĝo de fenilalanino kaj tirozino en la sango estas kontrolita, la agado de hepataj enzimoj (fenilalanina hidroksilase) estas determinita, biokemia ekzameno de urino (determino de ketonaj acidoj), metabolitoj de kateolaminoj en la urino, estas plenumata de infano kaj ekzameno de la infano.
Genetika difekto en fenilketonuria povas esti detektita eĉ dum gravedeco dum invasiva antaŭnaska diagnozo de la feto (korionbiopsio, amniocentesis, kordocentezo).
La diferenca diagnozo de fenilketonuria estas farata kun intrakrania naska traŭmato en novnaskitoj, intrauterinaj infektoj kaj aliaj metabolaj malordoj de aminoacidoj.
Traktado de Fenilketonuria
Fundamenta faktoro en la kuracado de fenilketonuria estas dieto, kiu limigas la konsumon de proteinoj en la korpo. Kuracado rekomendas komenci koncentriĝon de fenilalanino> 6 mg%. Specialaj miksaĵoj estis ellaboritaj por beboj - Afenilak, Lofenilak, por infanoj pli ol 1-jaraj - Tetrafen, Phenyl-free, pli ol 8-jaraj - Maxamum-XP kaj aliaj .La bazo de la dieto estas malaltaj proteinoj - fruktoj, legomoj, sukoj, proteinoj hidrolizataj kaj miksaĵoj de aminoacidoj. . Pliigo de la dieto eblas post 18 jaroj lige kun pliigo de toleremo al fenilalanino. Konforme al rusa juro, disponigado de medicina nutrado al homoj suferantaj de fenilketonuria devas esti senpaga.
Al pacientoj oni preskribas konsumon de mineralaj komponaĵoj, vitaminojn de la grupo B ktp., Laŭ indikoj - nootropaj drogoj, kontraŭkonvulsivaj. En la kompleksa terapio de fenilketonuria, ĝenerala masaĝo, ekzercoterapio kaj akupunkturo estas vaste uzataj.
Infanoj suferantaj de fenilketonuria estas sub la superrigardo de loka infankuracisto kaj neuropsikiatro kaj ofte bezonas la helpon de paroloterapeŭto kaj patologiisto. Zorgema monitorado de la neŭropsika stato de infanoj, monitorado de la nivelo de fenilalanino en la sango kaj elektroencefalogramoj estas necesaj.
Atipaj formoj de fenilcetonuria, kiuj ne taŭgas por dieta traktado, postulas nomumon de hepatoprotektoroj, kontraŭkonvulsivaj, anstataŭiga terapio kun levodopa, 5-hidroxitriptopano.
Antaŭdiro kaj antaŭzorgo de fenilketonuria
Efektivigi masan kribradon por fenilketonuria en la novnaska periodo permesas organizi fruan dietan terapion kaj antaŭvidi severan cerebran damaĝon, difektitan hepatan funkcion. Kun la frua nomumo de elimina dieto por klasika fenilketonuria, la prognozo por disvolviĝo de infanoj estas bona. Se kuracado komenciĝas malfrue, la prognozo por mensa disvolviĝo estas malbona.
Antaŭzorgo de komplikaĵoj de fenilketonuria konsistas en amasa kribrado de novnaskitoj, frua preskribado kaj longtempa konformiĝo al dieta nutrado.
Por taksi la riskon naski infanon kun fenilketonuria, antaŭdona genetika konsilado estu donita al paroj, kiuj jam havas malsanan infanon, estas en konsangula rilato kaj havas parencojn kun ĉi tiu malsano. Virinoj kun fenilketonuria plananta gravedecon devas sekvi striktan dieton antaŭ koncepto kaj dum gravedeco por ekskludi kreskon de la nivelo de fenilalanino kaj ĝiaj metabolitoj kaj malgravigitan disvolviĝon de genetike sana feto. La risko havi infanon kun fenilketonuria ĉe gepatroj portantaj difektitan genon estas 1: 4.




-
Sakarino estas la unua sekura dolĉigilo
Sakarino estas la unua sekura dolĉigilo Sakarino estas sekura sukero-anstataŭanto. Priskribo, avantaĝoj kaj kontraŭoj, kontraŭindikoj kaj uzo. Komparo kun fruktozo kaj sukralose. ... -

-

-